





Polymerization based on Green Chemistry
Current environmental constraints along with the depletion of fossil fuels necessitates the use of greener materials and processes in society. Thus, research on biorenewable plastics has tremendously increased over the past 20-30 years, now that the consequences of using oil based plastics unfold on the environment [1–3]. Not only is the focus placed on the recycling of already made plastics but also on the ‘green chemistry’ of producing new plastics from different biomass sources [4–6]. Green Chemistry is the collective term for a set of 12 principles meant to provide a guideline for a circular economy. This project is divided into 3 subprojects, focusing on biorenewable starting materials, recyclable materials, and retrieving unreacted chemicals.


Subproject 2: Blends of SBS triblock polymer with poly(Styrene-b-methylene butyrolactone)
Thermoplastic elastomers (TPE) are an interesting class of materials due to the fact that they possess both the properties of glassy/semi-crystalline thermoplastics and soft elastomers. Because of this biphasic nature, TPEs are essentially rubbery materials that are able to be processed as thermoplastics [28-31]. The properties of TPEs are thermally reversible making them ideally suited for high-throughput thermoplastic processes, such as melt extrusion and injection molding [32].
Generally, TPEs are composed of structured A-B-A block copolymers, where A is commonly a thermoplastic block polymer and B is an elastomeric block polymer. The final observable properties are mainly dependent on factors such as choice of monomers, block length, and the weight fractions of A and B. Due to thermodynamic incompatibility between the two components, they undergo microphase separation and form domain structures that are characterized by "hard" segment domains dispersed in a "soft" segment matrix. The "hard" domains can form a physical three-dimensional network due to the covalent bonds between the chemically dissimilar components. Consequently, they exhibit mechanical properties that are relatively comparable to those of vulcanized rubber [32].
An example of a TPE that is the focus of this project is polystyrene–polybutadiene–polystyrene block copolymer (SBS) , in which the molecular weights of polystyrene (PS) and polybutadiene (PB) and the weight fraction of PS are restricted to a certain quantity.
In our project SBS consists out of 30 wt% polystyrene and 70wt% polybutadiene. PB is the macro-domain in the matrix where PS is dispersed. Due to the fact that this rubber is a block polymer, two distinctive glass transition temperatures (Tg) are known. Around -100 Celsius for PB, and +100 Celsius for PS. The glass transition temperature is the temperature range where the polymer substrate changes from a rigid, glassy material to a soft (not melted) material. It's usually measured in terms of the stiffness, or modulus.
When the temperature is between the Tg of the blocks, the PB acts as soft chains that can move around freely, while the rigid PS act as bridges between PS domains of other chains. However, when you go above 100 Celsius, this physical network gets destroyed and the rubber loses its mechanical strength quickly [33].
To solve this problem, a new block copolymer was made consisting out of polystyrene and poly(methylene butyrolactone) - p(MBL) - and blended with SBS. From the previous project, it has been established that p(MBL) polymers have excellent heat resistant properties. This newly formed polymer blend (SBS + poly(S-β-MBL) is pressed into discs and bars for testing. By combining that polymer with current SBS, a new blend will be made that retains their mechanical properties at higher temperature.


Subproject 1: RAFT Polymerization of a Biorenewable/ Sustainable Monomer Via a Green Process
Biobased monomers are the most sustainable, renewable, abundant and ubiquitous biomass available in nature, as they are present in plants. Hence, one of those aspects is to use biorenewable monomers, like α-methylene-butyrolactones. Those biobased monomers are cyclic analogues of methyl methacrylate (MMA), containing an exocyclic butyrolactone ring. α-methylene- γ-butyrolactone monomer can be found in extremely small quantities in tulips, hence it is also called Tulipalin A. A better way to obtain methylene lactones is to synthesize them from itaconic acid, which is present and readily available from biomass sources like rice and corn [7]. A few studies have been performed on transforming itaconic acid into compounds similar to cyclic lactones [7–12]. The polymerization of these monomers will lead to plastics with advanced thermal properties. On average the glass transition temperature of these cyclic lactones is around 195 Celsius. For the sake of comparison, poly methylmethacrylate (PMMA) and polystyrene (PS) only have a glass transition temperature of around 100 Celsius, making them unsuitable for higher temperature applications. Additionally, polymers obtained from these monomers possess solvent and scratch resistance and advanced optical properties [7,14].
By employing reversible addition−fragmentation chain-transfer (RAFT) polymerization technique, the control over the polymerization can be achieved [13-14]. The molecular weight can be pre-determined by choosing the right amount of RAFT agent, monomer and initiator concentration, while maintaining chain end fidelity and a low polydispersity index (PDI). By reactivation of the chain end groups of the functional polymer, new monomers can be added to the already existing polymer (i.e. chain extension) to produce block-, graft- and star- (co)polymers [15-17]. However, many efforts in polymerizing this class of monomers still relies on the use of classical organic solvents with the corresponding, generally high, environmental impact. In this the use of a green solvent would ideally render the overall product and process to make it much more attractive from an environmental point of view. Among all possible choices, the use of supercritical fluids, and more specifically supercritical carbon dioxide (scCO2), as an alternative solvent or extraction medium has been on the rise the past decade [18-21].
One of the important aspects why CO2 is being used is the general low parameter criteria to realize supercritical conditions (P=73 bar and T= 31 Celsius) and the low-toxicity and non-flammable aspect. Supercritical CO2 is a unique fluid with high diffusivity where it can been seen as a hybrid between a gas and a liquid, meaning the density of a liquid and the viscosity of a gas. Above the critical point of CO2 it is possible to adjust the parameters for each specific process. Typically higher pressures are being used for the polymerization reaction (~ 300 bar) as that increases the solvation power of the scCO2, due to an increase of the density. RAFT polymerization of styrene and methyl methacrylate in scCO2 have already been extensively researched but, to best of our knowledge, the use of a biorenewable monomer for RAFT polymerization in scCO2 has not been reported yet [22–27].
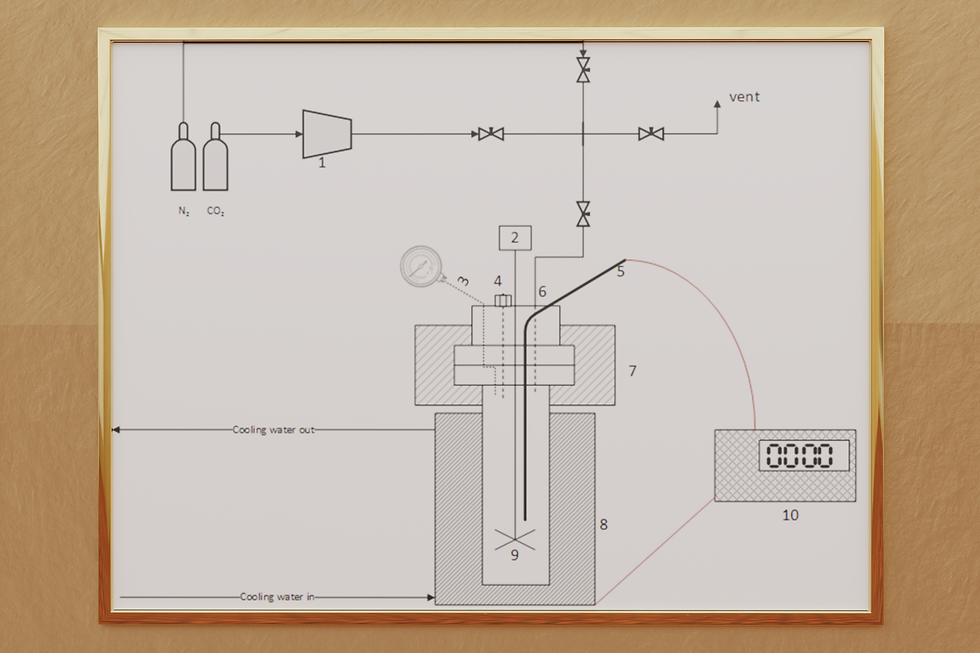
On the left you see a schematic of the reactor setup

This vacuum devolatilizer may be heated to 250-280 Celsius and coincidentally, this unit contributes to the highest emission of gVOC/kg product the plant produces. By using supercritical CO2 we plan on drastically reducing these emissions while maintaining high extraction yield of styrene from PS.

Subproject 3: Extraction of residual styrene monomer from bulk polymerized polystyrene using a continuous supercritical CO2 extractor
Polystyrene is the fourth largest thermoplastic by production volume. It is used in applications in the following major markets (listed in order of consumption): packaging, consumer/institutional goods, electrical/electronic goods, building/construction, furniture, industrial/machinery, and transportation.
Homopolymers and copolymers can be produced by bulk (or mass), solution (a modified bulk), suspension, or emulsion polymerization techniques. The bulk (mass) process is the most widely used process for polystyrene today. Bulk polymerization can happen in a continuous - or a batch process. What both systems have in common is the use of a vacuum devolatilizer to remove unreacted styrene and other impurities.
On the left you see a sample before and after extraction [34].
[1] Hoel, M.; Kverndokk, S. Depletion of Fossil Fuels and the Impacts of Global Warming. Resour. Energy Econ. 1996, 18 (2), 115–136. https://doi.org/10.1016/0928-7655(96)00005-X.
[2] Allen, L.; Cohen, M. J.; Abelson, D.; Miller, B. Fossil Fuels and Water Quality. The World’s Water 2012, 73–96. https://doi.org/10.5822/978-1-59726-228-6_4.
[3] Raddadi, N.; Fava, F. Biodegradation of Oil-Based Plastics in the Environment: Existing Knowledge and Needs of Research and Innovation. Sci. Total Environ. 2019, 679, 148–158. https://doi.org/10.1016/J.SCITOTENV.2019.04.419.
[4] Nonato, R. V.; Mantelatto, P. E.; Rossell, C. E. V. Integrated Production of Biodegradable Plastic, Sugar and Ethanol. Appl. Microbiol. Biotechnol. 2001 571 2001, 57 (1), 1–5. https://doi.org/10.1007/S002530100732.
[5] Wei, W.; Keh, C. C. K.; Li, C.-J.; Varma, R. S. Water as a Reaction Medium for Clean Chemical Processes. Clean Technol. Environ. Policy 2004, 6 (4), 250–257. https://doi.org/10.1007/s10098-003-0242-7.
[6]6 Hatton, F. L. Recent Advances in RAFT Polymerization of Monomers Derived from Renewable Resources. Polym. Chem. 2020, 11 (2), 220–229. https://doi.org/10.1039/C9PY01128E.
[7] Trotta, J. T.; Jin, M.; Stawiasz, K. J.; Michaudel, Q.; Chen, W. L.; Fors, B. P. Synthesis of Methylene Butyrolactone Polymers from Itaconic Acid. J. Polym. Sci. Part A Polym. Chem. 2017, 55 (17), 2730–2737. https://doi.org/10.1002/POLA.28654.
[8] Xu, S.; Huang, J.; Xu, S.; Luo, Y. RAFT Ab Initio Emulsion Copolymerization of γ-Methyl-α-Methylene-γ-Butyrolactone and Styrene. Polymer (Guildf). 2013, 54 (7), 1779–1785. https://doi.org/10.1016/J.POLYMER.2013.02.007.
[9] Qi, G.; Nolan, M.; Schork, F. J.; Jones, C. W. Emulsion and Controlled Miniemulsion Polymerization of the Renewable Monomer γ-Methyl-α-Methylene-γ-Butyrolactone. J. Polym. Sci. Part A Polym. Chem. 2008, 46 (17), 5929–5944. https://doi.org/10.1002/POLA.22909.
[10] Macromolecules, J. M. Atom Transfer Radical Polymerization of Tulipalin A: A Naturally Renewable Monomer. 2008. https://doi.org/10.1021/ma8010813.
[11] Mosnáček, J.; Yoon, J. A.; Juhari, A.; Koynov, K.; Matyjaszewski, K. Synthesis, Morphology and Mechanical Properties of Linear Triblock Copolymers Based on Poly(α-Methylene-γ-Butyrolactone). Polymer (Guildf). 2009, 50 (9), 2087–2094. https://doi.org/10.1016/J.POLYMER.2009.02.037.
[12] Green, D. C.; Allen, K. W.; Chem Soc, J.; Commun, C.; Kaplan, M. L.; Haddon, R. C.; Wudl, F.; Feit, E. D.; Org, J.; Pittman, U.; Patterson, W. J.; McManus, S. P.; Polym Sci, J.; Akkapeddi, M. K. Poly(α-Methylene-γ-Butyrolactone) Synthesis, Configurational Structure, and Properties. Macromolecules 2002, 12 (4), 546–551. https://doi.org/10.1021/MA60070A002.
[13] Fetizon, M.; Golfier, M.; Louis, J. M. Oxydations Par Le Carbonate d’argent Sur Celite—XIII : Preparation de Lactones. Tetrahedron 1975, 31 (2), 171–176. https://doi.org/10.1016/0040-4020(75)85013-7.
[14] Trotta, J. T. Renewable Polymers from Itaconic Acid. 2019. https://doi.org/10.7298/CAH5-HX28.
[15] Moad, G.; Chiefari, J.; Chong, Y.; Krstina, J.; Mayadunne, R. T.; Postma, A.; Rizzardo, E.; Thang, S. H. Living Free Radical Polymerization with Reversible Addition-Fragmentation Chain Transfer (the Life of RAFT). https://doi.org/10.1002/1097-0126.
[16] Chiefari, J.; Chong, Y. K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T. P. T.; Mayadunne, R. T. A.; Meijs, G. F.; Moad, C. L.; Moad, G.; Rizzardo, E.; Thang, S. H. Living Free-Radical Polymerization by Reversible Addition−Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31 (16), 5559–5562. https://doi.org/10.1021/MA9804951.
References
[17] Chong, Y. K.; T Le, T. P.; Moad, G.; Rizzardo, E.; Thang, S. H. A More Versatile Route to Block Copolymers and Other Polymers of Complex Architecture by Living Radical Polymerization: The RAFT Process. 1999. https://doi.org/10.1021/ma981472p.
[18] Barner, L.; Christopher Barner-Kowollik, B.; Davis, T. P.; Stenzel, M. H. Complex Molecular Architecture Polymers via RAFT. 2004. https://doi.org/10.1071/CH03232.
[19] Moad, G.; Rizzardo, E.; Thang, S. H. RAFT Polymerization and Some of Its Applications. Chem. – An Asian J. 2013, 8 (8), 1634–1644. https://doi.org/10.1002/ASIA.201300262.
[20] Peker, H.; Srinivasan, M. P.; Smith, J. M.; McCoy, B. J. Caffeine Extraction Rates from Coffee Beans with Supercritical Carbon Dioxide. AIChE J. 1992, 38 (5), 761–770. https://doi.org/10.1002/AIC.690380513.
[21] DeSimone, J. M.; Guan, Z.; Elsbernd, C. S. Synthesis of Fluoropolymers in Supercritical Carbon Dioxide. Science (80-. ). 1992, 257 (5072), 945–947. https://doi.org/10.1126/SCIENCE.257.5072.945.
[22] Nalawade, S. P.; Picchioni, F.; Janssen, L. P. B. M. Supercritical Carbon Dioxide as a Green Solvent for Processing Polymer Melts: Processing Aspects and Applications. Prog. Polym. Sci. 2006, 31 (1), 19–43. https://doi.org/10.1016/J.PROGPOLYMSCI.2005.08.002.
[24] Kendall, J. L.; Canelas, D. A.; Young, J. L.; Desimone, J. M. Polymerizations in Supercritical Carbon Dioxide. 1999. https://doi.org/10.1021/cr9700336.
[24] Gregory, A. M.; Thurecht, K. J.; Howdle, S. M. Controlled Dispersion Polymerization of Methyl Methacrylate in Supercritical Carbon Dioxide via RAFT. Macromolecules 2008, 41 (4), 1215–1222. https://doi.org/10.1021/MA702017R/SUPPL_FILE/MA702017R-FILE001.PDF.
[25] Thurecht, K. J.; Gregory, A. M.; Wang, W.; Howdle, S. M. “Living” Polymer Beads in Supercritical CO2. Macromolecules 2007, 40 (9), 2965–2967. https://doi.org/10.1021/MA062724M/SUPPL_FILE/MA062724MSI20061127_121524.PDF.
[26] Arita, T.; Beuermann, S.; Buback, M.; Vana, P. RAFT Polymerization of Methyl Acrylate in Carbon Dioxide. Macromol. Mater. Eng. 2005, 290 (4), 283–293. https://doi.org/10.1002/MAME.200400274.
[27] Arita, T.; Beuermann, S.; Buback, M.; Vana, P. Reversible Addition Fragmentation Chain Transfer (RAFT) Polymerization of Styrene in Fluid CO2. E-Polymers 2004, 4 (1), 1–14. https://doi.org/10.1515/EPOLY.2004.4.1.20/MACHINEREADABLECITATION/RIS.
[28] Gabriëlse, W., Soliman, M., and Dijkstra, K. (2001). Microstructure and phase behavior of block copoly (ether ester) thermoplastic elastomers. Macromolecules, 34(6):1685–1693.
[29] Spontak, R. J. and Patel, N. P. (2000). Thermoplastic elastomers: fundamentals and applications. Current opinion in colloid & interface science, 5(5-6):333–340.
[30] Abdou-Sabet, S., Puydak, R., and Rader, C. (1996). Dynamically vulcanized thermoplastic elastomers. Rubber chemistry and technology, 69(3):476–494.
[31] Müller, G. and Rieger, B. (2002). Propene based thermoplastic elastomers by early and late transition metal catalysis. Progress in polymer science, 27(5):815–851.
[32] Holden, G. (1987). Thermoplastic elastomers. In Rubber technology, pages 465–481. Springer
[33] Picchioni, F., Casentini, E., Passaglia, E., & Ruggeri, G. (2003). Blends of SBS Triblock Copolymer with Poly(2,6-dimethyl-1,4-phenylene oxide)/Polystyrene Mixture.
[34] 6.6.3 Polystyrene 1-2. (n.d.).